Beginner’s Guide
Flan is a trace impurity transport code that is run on top of a pre-computed plasma background. As of now, that is provided by the gyrokinetic solver within Gkeyll. It is certainly beyond the scope of this guide to detail how to use Gkeyll, so instead we will walk through the steps used to obtain a generic Gkeyll simulation representative of the DIII-D far-SOL assuming you already have a working Gkeyll installation.
If one is familiar with trace impurity transport codes like DIVIMP or ERO2.0, then one can think of the relationship between Gkeyll and Flan as similar to that between, say, SOLPS-ITER and ERO2.0. SOLPS-ITER is used to generate a (2D) background that is then used in ERO2.0 to follow a trace impurity like tungsten. Same idea, except Gkeyll provides the background plasma to Flan. Flan is more tightly coupled to Gkeyll though, in that it uses the same computational grid as Gkeyll. This facilitates coupling, and enables rather efficient simulations due to simplifications in translating between physical space and the computational field-aligned space of Gkeyll with the reciprocal basis vectors (this is an implementation detail, do not worry too much about it at this point).
Generating a Plasma Background with Gkeyll
This section provides an Gkeyll input file that generates a turbulent representation of a generic DIII-D far-SOL, #167196. The simulation has been benchmarked against experimental reciprocating Langmuir probe ne and Te data. See Zamperini, S. A., et al. Nucl. Fusion 64, 074002 (2024) for more details (this was actually for a prototype of Flan, but the background is more or less the same as what is supplied below).
Gkeyll Input File
The below code is a Gkeyll input file for this guide. This was compiled and ran on the Perlmutter supercomputer at NERSC for about 2.5 days using 8 GPUs. The Gkeyll commit used for this simulation was eb9c458f27f0.
#include <math.h>
#include <stdio.h>
#include <stdlib.h>
#include <time.h>
#include <gkyl_alloc.h>
#include <gkyl_const.h>
#include <gkyl_gyrokinetic.h>
#include <gkyl_util.h>
#include <rt_arg_parse.h>
struct sheath_ctx
{
int cdim, vdim; // Dimensionality.
// Physical constants (using non-normalized physical units).
double epsilon0; // Permittivity of free space.
double mass_elc; // Electron mass.
double charge_elc; // Electron charge.
double mass_ion; // Proton mass.
double charge_ion; // Proton charge.
double Te; // Electron temperature.
double Ti; // Ion temperature.
double n0; // Reference number density (1 / m^3).
double B_axis; // Magnetic field axis (simple toroidal coordinates).
double R0; // Major radius (simple toroidal coordinates).
double a0; // Minor axis (simple toroidal coordinates).
double nu_frac; // Collision frequency fraction.
// Derived physical quantities (using non-normalized physical units).
double R; // Radial coordinate (simple toroidal coordinates).
double B0; // Reference magnetic field strength (Tesla).
double log_lambda_elc; // Electron Coulomb logarithm.
double log_lambda_ion; // Ion Coulomb logarithm.
double nu_elc; // Electron collision frequency.
double nu_ion; // Ion collision frequency.
double c_s; // Sound speed.
double vte; // Electron thermal velocity.
double vti; // Ion thermal velocity.
double omega_ci; // Ion cyclotron frequency.
double rho_s; // Ion-sound gyroradius.
double n_src; // Source number density.
double T_src; // Source temperature.
double xmu_src; // Source mean position (x-direction).
double xsigma_src; // Source standard deviation (x-direction).
double floor_src; // Minimum source intensity.
// Simulation parameters.
int Nx; // Cell count (configuration space: x-direction).
int Ny; // Cell count (configuration space: y-direction).
int Nz; // Cell count (configuration space: z-direction).
int Nvpar; // Cell count (velocity space: parallel velocity direction).
int Nmu; // Cell count (velocity space: magnetic moment direction).
int cells[GKYL_MAX_DIM]; // Number of cells in all directions.
double Lx; // Domain size (configuration space: x-direction).
double Ly; // Domain size (configuration space: y-direction).
double Lz; // Domain size (configuration space: z-direction).
double vpar_max_elc; // Domain boundary (electron velocity space: parallel velocity direction).
double mu_max_elc; // Domain boundary (electron velocity space: magnetic moment direction).
double vpar_max_ion; // Domain boundary (ion velocity space: parallel velocity direction).
double mu_max_ion; // Domain boundary (ion velocity space: magnetic moment direction).
int poly_order; // Polynomial order.
double cfl_frac; // CFL coefficient.
double t_end; // Final simulation time.
int num_frames; // Number of output frames.
double write_phase_freq; // Frequency of writing phase-space diagnostics (as a fraction of num_frames).
int int_diag_calc_num; // Number of integrated diagnostics computations (=INT_MAX for every step).
double dt_failure_tol; // Minimum allowable fraction of initial time-step.
int num_failures_max; // Maximum allowable number of consecutive small time-steps.
};
struct sheath_ctx
create_ctx(void)
{
int cdim = 3, vdim = 2; // Dimensionality.
// Physical constants (using non-normalized physical units).
double epsilon0 = GKYL_EPSILON0; // Permittivity of free space.
double mass_elc = GKYL_ELECTRON_MASS; // Electron mass.
double mass_ion = 2.014 * GKYL_PROTON_MASS; // Proton mass.
double charge_elc = -GKYL_ELEMENTARY_CHARGE; // Electron charge.
double charge_ion = GKYL_ELEMENTARY_CHARGE; // Proton charge.
double eV = GKYL_ELEMENTARY_CHARGE;
double Te = 15.0 * GKYL_ELEMENTARY_CHARGE; // Electron temperature.
double Ti = 15.0 * GKYL_ELEMENTARY_CHARGE; // Ion temperature.
double n0 = 7.0e18; // Reference number density (1 / m^3).
double B_axis = 2.04; // Magnetic field axis (simple toroidal coordinates).
double R0 = 1.722; // Major radius (simple toroidal coordinates).
double a0 = 0.59; // Minor axis (simple toroidal coordinates).
double nu_frac = 0.1; // Collision frequency fraction.
// Derived physical quantities (using non-normalized physical units).
//double R = R0 + a0; // Radial coordinate (simple toroidal coordinates).
double R = 2.30;
double B0 = B_axis * (R0 / R); // Reference magnetic field strength (Tesla).
double log_lambda_elc = 6.6 - 0.5 * log(n0 / 1.0e20) + 1.5 * log(Te / charge_ion); // Electron Coulomb logarithm.
double log_lambda_ion = 6.6 - 0.5 * log(n0 / 1.0e20) + 1.5 * log(Ti / charge_ion); // Ion Coulomb logarithm.
double nu_elc = nu_frac * log_lambda_elc * pow(charge_ion, 4.0) * n0 /
(6.0 * sqrt(2.0) * pow(M_PI, 3.0 / 2.0) * pow(epsilon0, 2.0) * sqrt(mass_elc) * pow(Te, 3.0 / 2.0)); // Electron collision frequency.
double nu_ion = nu_frac * log_lambda_ion * pow(charge_ion, 4.0) * n0 /
(12.0 * pow(M_PI, 3.0 / 2.0) * pow(epsilon0, 2.0) * sqrt(mass_ion) * pow(Ti, 3.0 / 2.0)); // Ion collision frequency.
double c_s = sqrt(Te / mass_ion); // Sound speed.
double vte = sqrt(Te / mass_elc); // Electron thermal velocity.
double vti = sqrt(Ti / mass_ion); // Ion thermal velocity.
double omega_ci = fabs(charge_ion * B0 / mass_ion); // Ion cyclotron frequency.
double rho_s = c_s / omega_ci; // Ion-sound gyroradius.
double n_src = 1.4690539 * 3.612270e23 * 0.10; // Source number density.
//double T_src = 2.0 * Te; // Source temperature.
double T_src = 50.0 * eV;
double xmu_src = R + 0.005; // Source mean position (x-direction).
double xsigma_src = 0.005; // Source standard deviation (x-direction).
double floor_src = 0.1; // Minimum source intensity.
// Simulation parameters.
int Nx = 48; // Cell count (configuration space: x-direction).
int Ny = 32; // Cell count (configuration space: y-direction).
int Nz = 8; // Cell count (configuration space: z-direction).
int Nvpar = 10; // Cell count (velocity space: parallel velocity direction).
int Nmu = 5; // Cell count (velocity space: magnetic moment direction).
double Lx = 150.0 * rho_s; // Domain size (configuration space: x-direction).
double Ly = 100.0 * rho_s; // Domain size (configuration space: y-direction).
double Lz = 10.0; // Domain size (configuration space: z-direction).
double vpar_max_elc = 4.0 * vte; // Domain boundary (electron velocity space: parallel velocity direction).
double mu_max_elc = (3.0 / 2.0) * 0.5 * mass_elc * pow(4.0 * vte, 2.0) / (2.0 * B0); // Domain boundary (electron velocity space: magnetic moment direction).
double vpar_max_ion = 4.0 * vti; // Domain boundary (ion velocity space: parallel velocity direction).
double mu_max_ion = (3.0 / 2.0) * 0.5 * mass_ion * pow(4.0 * vti, 2.0) / (2.0 * B0); // Domain boundary (ion velocity space: magnetic moment direction).
int poly_order = 1; // Polynomial order.
double cfl_frac = 0.50; // CFL coefficient.
//double t_end = 6.0e-6; // Final simulation time.
double t_end = 0.5e-3;
int num_frames = 1000; // Number of output frames.
double write_phase_freq = 0.2; // Frequency of writing phase-space diagnostics (as a fraction of num_frames).
int int_diag_calc_num = num_frames*100;
double dt_failure_tol = 1.0e-4; // Minimum allowable fraction of initial time-step.
int num_failures_max = 20; // Maximum allowable number of consecutive small time-steps.
struct sheath_ctx ctx = {
.cdim = cdim,
.vdim = vdim,
.epsilon0 = epsilon0,
.mass_elc = mass_elc,
.charge_elc = charge_elc,
.mass_ion = mass_ion,
.charge_ion = charge_ion,
.Te = Te,
.Ti = Ti,
.n0 = n0,
.B_axis = B_axis,
.R0 = R0,
.a0 = a0,
.nu_frac = nu_frac,
.R = R,
.B0 = B0,
.log_lambda_elc = log_lambda_elc,
.nu_elc = nu_elc,
.log_lambda_ion = log_lambda_ion,
.nu_ion = nu_ion,
.c_s = c_s,
.vte = vte,
.vti = vti,
.omega_ci = omega_ci,
.rho_s = rho_s,
.n_src = n_src,
.T_src = T_src,
.xmu_src = xmu_src,
.xsigma_src = xsigma_src,
.floor_src = floor_src,
.Nx = Nx,
.Ny = Ny,
.Nz = Nz,
.Nvpar = Nvpar,
.Nmu = Nmu,
.cells = {Nx, Ny, Nz, Nvpar, Nmu},
.Lx = Lx,
.Ly = Ly,
.Lz = Lz,
.vpar_max_elc = vpar_max_elc,
.mu_max_elc = mu_max_elc,
.vpar_max_ion = vpar_max_ion,
.mu_max_ion = mu_max_ion,
.poly_order = poly_order,
.cfl_frac = cfl_frac,
.t_end = t_end,
.num_frames = num_frames,
.write_phase_freq = write_phase_freq,
.int_diag_calc_num = int_diag_calc_num,
.dt_failure_tol = dt_failure_tol,
.num_failures_max = num_failures_max,
};
return ctx;
}
void
evalElcDensityInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0], z = xn[2];
double mass_ion = app->mass_ion;
double n_src = app->n_src;
double T_src = app->T_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double floor_src = app->floor_src;
double Lz = app->Lz;
double src_density = GKYL_MAX2(exp(-((x - xmu_src) * (x - xmu_src)) / ((2.0 * xsigma_src) * (2.0 * xsigma_src))), floor_src) * n_src;
double src_temp = 0.0;
double n = 0;
if (x < xmu_src + 3.0 * xsigma_src) {
src_temp = T_src;
}
else {
src_temp = (3.0 / 8.0) * T_src;
}
double c_s_src = sqrt((5.0 / 3.0) * src_temp / mass_ion);
double n_peak = 4.0 * sqrt(5.0) / 3.0 / c_s_src * (0.125 * Lz) * src_density;
if (fabs(z) <= 0.25 * Lz) {
n = 0.5 * n_peak * (1.0 + sqrt(1.0 - (z / (0.25 * Lz)) * (z / (0.25 * Lz)))); // Electron total number density (left).
}
else {
n = 0.5 * n_peak; // Electron total number density (right).
}
// Set electron total number density.
fout[0] = n;
}
void
evalElcTempInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0];
double Te = app->Te;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double T = 0.0;
if (x < xmu_src + 3.0 * xsigma_src) {
T = (5.0 / 4.0) * Te; // Electron isotropic temperature (left).
}
else {
T = 0.5 * Te; // Electron isotropic temperature (right).
}
// Set electron isotropic temperature.
fout[0] = T;
}
void
evalElcUparInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
// Set electron parallel velocity.
fout[0] = 0.0;
}
void
evalElcSourceDensityInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0], z = xn[2];
double n_src = app->n_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double floor_src = app->floor_src;
double Lz = app->Lz;
double n = 0.0;
if (fabs(z) < 0.25 * Lz) {
n = GKYL_MAX2(exp(-((x - xmu_src) * (x - xmu_src)) / ((2.0 * xsigma_src) * (2.0 * xsigma_src))),
floor_src) * n_src; // Electron source total number density (left).
}
else {
n = 1.0e-40 * n_src; // Electron source total number density (right).
}
// Set electron source total number density.
fout[0] = n;
}
void
evalElcSourceTempInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0];
double T_src = app->T_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double T = 0.0;
if (x < xmu_src + 3.0 * xsigma_src) {
T = T_src; // Electron source isotropic temperature (left).
}
else {
T = (3.0 / 8.0) * T_src; // Electron source isotropic temperature (right).
}
// Set electron source isotropic temperature.
fout[0] = T;
}
void
evalElcSourceUparInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
// Set electron source parallel velocity.
fout[0] = 0.0;
}
void
evalIonDensityInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0], z = xn[2];
double mass_ion = app->mass_ion;
double n_src = app->n_src;
double T_src = app->T_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double floor_src = app->floor_src;
double Lz = app->Lz;
double src_density = GKYL_MAX2(exp(-((x - xmu_src) * (x - xmu_src)) / ((2.0 * xsigma_src) * (2.0 * xsigma_src))), floor_src) * n_src;
double src_temp = 0.0;
double n = 0;
if (x < xmu_src + 3.0 * xsigma_src) {
src_temp = T_src;
}
else {
src_temp = (3.0 / 8.0) * T_src;
}
double c_s_src = sqrt((5.0 / 3.0) * src_temp / mass_ion);
double n_peak = 4.0 * sqrt(5.0) / 3.0 / c_s_src * (0.125 * Lz) * src_density;
if (fabs(z) <= 0.25 * Lz) {
n = 0.5 * n_peak * (1.0 + sqrt(1.0 - (z / (0.25 * Lz)) * (z / (0.25 * Lz)))); // Ion total number density (left).
}
else {
n = 0.5 * n_peak; // Ion total number density (right).
}
// Set ion total number density.
fout[0] = n;
}
void
evalIonTempInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0];
double Ti = app->Ti;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double T = 0.0;
if (x < xmu_src + 3.0 * xsigma_src) {
T = (5.0 / 4.0) * Ti; // Ion isotropic temperature (left).
}
else {
T = 0.5 * Ti; // Ion isotropic temperature (right).
}
// Set ion isotropic temperature.
fout[0] = T;
}
void
evalIonUparInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
// Set ion parallel velocity.
fout[0] = 0.0;
}
void
evalIonSourceDensityInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0], z = xn[2];
double n_src = app->n_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double floor_src = app->floor_src;
double Lz = app->Lz;
double n = 0.0;
if (fabs(z) < 0.25 * Lz) {
n = GKYL_MAX2(exp(-((x - xmu_src) * (x - xmu_src)) / ((2.0 * xsigma_src) * (2.0 * xsigma_src))),
floor_src) * n_src; // Ion source total number density (left).
}
else {
n = 1.0e-40 * n_src; // Ion source total number density (right).
}
// Set ion source total number density.
fout[0] = n;
}
void
evalIonSourceTempInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = xn[0];
double T_src = app->T_src;
double xmu_src = app->xmu_src;
double xsigma_src = app->xsigma_src;
double T = 0.0;
if (x < xmu_src + 3.0 * xsigma_src) {
T = T_src; // Ion source isotropic temperature (left).
}
else {
T = (3.0 / 8.0) * T_src; // Ion source isotropic temperature (right).
}
// Set ion source isotropic temperature.
fout[0] = T;
}
void
evalIonSourceUparInit(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
// Set ion source parallel velocity.
fout[0] = 0.0;
}
void
evalElcNu(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double nu_elc = app->nu_elc;
// Set electron collision frequency.
fout[0] = nu_elc;
}
void
evalIonNu(double t, const double* GKYL_RESTRICT xn, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double nu_ion = app->nu_ion;
// Set ion collision frequency.
fout[0] = nu_ion;
}
static inline void
mapc2p(double t, const double* GKYL_RESTRICT zc, double* GKYL_RESTRICT xp, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = zc[0], y = zc[1], z = zc[2];
double R0 = app->R0;
double a0 = app->a0;
double R = x;
double phi = z / (R0 + a0);
double X = R * cos(phi);
double Y = R * sin(phi);
double Z = y;
// Set physical coordinates (X, Y, Z) from computational coordinates (x, y, z).
// SAZ: The regression file for some reason had these lines swapped. I am
// not sure why they would do that, so I swapped them out.
xp[0] = X; xp[1] = Y; xp[2] = Z;
//xp[0] = x; xp[1] = y; xp[2] = z;
}
void
bmag_func(double t, const double* GKYL_RESTRICT zc, double* GKYL_RESTRICT fout, void* ctx)
{
struct sheath_ctx *app = ctx;
double x = zc[0];
double B0 = app->B0;
double R = app->R;
// Set magnetic field strength.
// SAZ: The regression file for some reason had these lines swapped. I am
// not sure why they would do that, so I swapped them out.
fout[0] = B0 * R / x;
//fout[0] = B0;
}
void
calc_integrated_diagnostics(struct gkyl_tm_trigger* iot, gkyl_gyrokinetic_app* app,
double t_curr, bool is_restart_IC, bool force_calc, double dt)
{
if (!is_restart_IC && (gkyl_tm_trigger_check_and_bump(iot, t_curr) || force_calc)) {
gkyl_gyrokinetic_app_calc_field_energy(app, t_curr);
gkyl_gyrokinetic_app_calc_integrated_mom(app, t_curr);
if ( !(dt < 0.0) )
gkyl_gyrokinetic_app_save_dt(app, t_curr, dt);
}
}
void
write_data(struct gkyl_tm_trigger* iot_conf, struct gkyl_tm_trigger* iot_phase,
gkyl_gyrokinetic_app* app, double t_curr, bool is_restart_IC, bool force_write)
{
bool trig_now_conf = gkyl_tm_trigger_check_and_bump(iot_conf, t_curr);
if (trig_now_conf || force_write) {
int frame = (!trig_now_conf) && force_write? iot_conf->curr : iot_conf->curr-1;
gkyl_gyrokinetic_app_write_conf(app, t_curr, frame);
if (!is_restart_IC) {
gkyl_gyrokinetic_app_write_field_energy(app);
gkyl_gyrokinetic_app_write_integrated_mom(app);
gkyl_gyrokinetic_app_write_dt(app);
}
}
bool trig_now_phase = gkyl_tm_trigger_check_and_bump(iot_phase, t_curr);
if (trig_now_phase || force_write) {
int frame = (!trig_now_conf) && force_write? iot_conf->curr : iot_conf->curr-1;
gkyl_gyrokinetic_app_write_phase(app, t_curr, frame);
}
}
int
main(int argc, char **argv)
{
struct gkyl_app_args app_args = parse_app_args(argc, argv);
#ifdef GKYL_HAVE_MPI
if (app_args.use_mpi) MPI_Init(&argc, &argv);
#endif
if (app_args.trace_mem) {
gkyl_cu_dev_mem_debug_set(true);
gkyl_mem_debug_set(true);
}
struct sheath_ctx ctx = create_ctx(); // Context for init functions.
int cells_x[ctx.cdim], cells_v[ctx.vdim];
for (int d=0; d<ctx.cdim; d++)
cells_x[d] = APP_ARGS_CHOOSE(app_args.xcells[d], ctx.cells[d]);
for (int d=0; d<ctx.vdim; d++)
cells_v[d] = APP_ARGS_CHOOSE(app_args.vcells[d], ctx.cells[ctx.cdim+d]);
// Construct communicator for use in app.
struct gkyl_comm *comm = gkyl_gyrokinetic_comms_new(app_args.use_mpi, app_args.use_gpu, stderr);
// Electrons.
struct gkyl_gyrokinetic_species elc = {
.name = "elc",
.charge = ctx.charge_elc, .mass = ctx.mass_elc,
.lower = { -ctx.vpar_max_elc, 0.0 },
.upper = { ctx.vpar_max_elc, ctx.mu_max_elc },
.cells = { cells_v[0], cells_v[1] },
.polarization_density = ctx.n0,
.projection = {
.proj_id = GKYL_PROJ_MAXWELLIAN_PRIM,
.density = evalElcDensityInit,
.ctx_density = &ctx,
.temp = evalElcTempInit,
.ctx_temp = &ctx,
.upar = evalElcUparInit,
.ctx_upar = &ctx,
},
.collisions = {
.collision_id = GKYL_LBO_COLLISIONS,
.self_nu = evalElcNu,
.ctx = &ctx,
.num_cross_collisions = 1,
.collide_with = { "ion" },
.normNu = true,
.n_ref = ctx.n0,
.T_ref = ctx.Te,
},
.source = {
.source_id = GKYL_PROJ_SOURCE,
.num_sources = 1,
.projection[0] = {
.proj_id = GKYL_PROJ_MAXWELLIAN_PRIM,
.density = evalElcSourceDensityInit,
.ctx_density = &ctx,
.temp = evalElcSourceTempInit,
.ctx_temp = &ctx,
.upar = evalElcSourceUparInit,
.ctx_upar = &ctx,
},
.diagnostics = {
.num_diag_moments = 1,
//.diag_moments = { GKYL_F_MOMENT_M0M1M2PARM2PERP },
.diag_moments = { GKYL_F_MOMENT_BIMAXWELLIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
// .time_integrated = true,
}
},
.bcx = {
.lower = { .type = GKYL_SPECIES_ZERO_FLUX, },
.upper = { .type = GKYL_SPECIES_ZERO_FLUX, },
},
.bcz = {
.lower = { .type = GKYL_SPECIES_GK_SHEATH, },
.upper = { .type = GKYL_SPECIES_GK_SHEATH, },
},
.num_diag_moments = 1,
//.diag_moments = { GKYL_F_MOMENT_M0, GKYL_F_MOMENT_M1, GKYL_F_MOMENT_M2, GKYL_F_MOMENT_M2PAR, GKYL_F_MOMENT_M2PERP },
.diag_moments = { GKYL_F_MOMENT_BIMAXWELLIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
.time_rate_diagnostics = true,
.boundary_flux_diagnostics = {
.num_diag_moments = 1,
.diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
// .time_integrated = true,
},
};
// Ions.
struct gkyl_gyrokinetic_species ion = {
.name = "ion",
.charge = ctx.charge_ion, .mass = ctx.mass_ion,
.lower = { -ctx.vpar_max_ion, 0.0 },
.upper = { ctx.vpar_max_ion, ctx.mu_max_ion },
.cells = { cells_v[0], cells_v[1] },
.polarization_density = ctx.n0,
.projection = {
.proj_id = GKYL_PROJ_MAXWELLIAN_PRIM,
.density = evalIonDensityInit,
.ctx_density = &ctx,
.temp = evalIonTempInit,
.ctx_temp = &ctx,
.upar = evalIonUparInit,
.ctx_upar = &ctx,
},
.collisions = {
.collision_id = GKYL_LBO_COLLISIONS,
.self_nu = evalIonNu,
.ctx = &ctx,
.num_cross_collisions = 1,
.collide_with = { "elc" },
.normNu = true,
.n_ref = ctx.n0,
.T_ref = ctx.Ti,
},
.source = {
.source_id = GKYL_PROJ_SOURCE,
.num_sources = 1,
.projection[0] = {
.proj_id = GKYL_PROJ_MAXWELLIAN_PRIM,
.density = evalIonSourceDensityInit,
.ctx_density = &ctx,
.temp = evalIonSourceTempInit,
.ctx_temp = &ctx,
.upar = evalIonSourceUparInit,
.ctx_upar = &ctx,
},
.diagnostics = {
.num_diag_moments = 1,
//.diag_moments = { GKYL_F_MOMENT_M0M1M2PARM2PERP },
.diag_moments = { GKYL_F_MOMENT_BIMAXWELLIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
// .time_integrated = true,
}
},
.bcx = {
.lower = { .type = GKYL_SPECIES_ZERO_FLUX, },
.upper = { .type = GKYL_SPECIES_ZERO_FLUX, },
},
.bcz = {
.lower = { .type = GKYL_SPECIES_GK_SHEATH, },
.upper = { .type = GKYL_SPECIES_GK_SHEATH, },
},
.num_diag_moments = 1,
//.diag_moments = { GKYL_F_MOMENT_M0M1M2PARM2PERP },
.diag_moments = { GKYL_F_MOMENT_BIMAXWELLIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
.time_rate_diagnostics = true,
.boundary_flux_diagnostics = {
.num_diag_moments = 1,
.diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
.num_integrated_diag_moments = 1,
.integrated_diag_moments = { GKYL_F_MOMENT_HAMILTONIAN },
// .time_integrated = true,
},
};
// Field.
struct gkyl_gyrokinetic_field field = {
.poisson_bcs = {
.lo_type = { GKYL_POISSON_DIRICHLET, GKYL_POISSON_PERIODIC },
.up_type = { GKYL_POISSON_DIRICHLET, GKYL_POISSON_PERIODIC },
.lo_value = { 0.0 },
.up_value = { 0.0 },
},
.time_rate_diagnostics = true,
};
// Gyrokinetic app.
struct gkyl_gk app_inp = {
.name = "d3d-167196-v9",
.cdim = ctx.cdim, .vdim = ctx.vdim,
//.lower = { ctx.R - (0.5 * ctx.Lx), -0.5 * ctx.Ly, -0.5 * ctx.Lz },
//.upper = { ctx.R + (0.5 * ctx.Lx), 0.5 * ctx.Ly, 0.5 * ctx.Lz },
.lower = { ctx.R, -0.5 * ctx.Ly, -0.5 * ctx.Lz },
.upper = { ctx.R + ctx.Lx, 0.5 * ctx.Ly, 0.5 * ctx.Lz },
.cells = { cells_x[0], cells_x[1], cells_x[2] },
.poly_order = ctx.poly_order,
.basis_type = app_args.basis_type,
.cfl_frac = ctx.cfl_frac,
// .cfl_frac_omegaH = 1e10,
.geometry = {
.geometry_id = GKYL_MAPC2P,
.world = { },
.mapc2p = mapc2p,
.c2p_ctx = &ctx,
.bmag_func = bmag_func,
.bmag_ctx = &ctx
},
.num_periodic_dir = 1,
.periodic_dirs = { 1 },
.num_species = 2,
.species = { elc, ion },
.field = field,
.parallelism = {
.use_gpu = app_args.use_gpu,
.cuts = { app_args.cuts[0], app_args.cuts[1], app_args.cuts[2] },
.comm = comm,
},
};
// Create app object.
gkyl_gyrokinetic_app *app = gkyl_gyrokinetic_app_new(&app_inp);
double t_curr = 0.0, t_end = ctx.t_end; // Initial and final simulation times.
int frame_curr = 0; // Initialize simulation.
if (app_args.is_restart) {
struct gkyl_app_restart_status status = gkyl_gyrokinetic_app_read_from_frame(app, app_args.restart_frame);
if (status.io_status != GKYL_ARRAY_RIO_SUCCESS) {
gkyl_gyrokinetic_app_cout(app, stderr, "*** Failed to read restart file! (%s)\n", gkyl_array_rio_status_msg(status.io_status));
goto freeresources;
}
frame_curr = status.frame;
t_curr = status.stime;
gkyl_gyrokinetic_app_cout(app, stdout, "Restarting from frame %d", frame_curr);
gkyl_gyrokinetic_app_cout(app, stdout, " at time = %g\n", t_curr);
}
else {
gkyl_gyrokinetic_app_apply_ic(app, t_curr);
}
// Create triggers for IO.
int num_frames = ctx.num_frames, num_int_diag_calc = ctx.int_diag_calc_num;
struct gkyl_tm_trigger trig_write_conf = { .dt = t_end/num_frames, .tcurr = t_curr, .curr = frame_curr };
struct gkyl_tm_trigger trig_write_phase = { .dt = t_end/(ctx.write_phase_freq*num_frames), .tcurr = t_curr, .curr = frame_curr};
struct gkyl_tm_trigger trig_calc_intdiag = { .dt = t_end/GKYL_MAX2(num_frames, num_int_diag_calc),
.tcurr = t_curr, .curr = frame_curr };
// Write out ICs (if restart, it overwrites the restart frame).
calc_integrated_diagnostics(&trig_calc_intdiag, app, t_curr, app_args.is_restart, false, -1.0);
write_data(&trig_write_conf, &trig_write_phase, app, t_curr, app_args.is_restart, false);
// Compute initial guess of maximum stable time-step.
double dt = t_end - t_curr;
// Initialize small time-step check.
double dt_init = -1.0, dt_failure_tol = ctx.dt_failure_tol;
int num_failures = 0, num_failures_max = ctx.num_failures_max;
long step = 1;
while ((t_curr < t_end) && (step <= app_args.num_steps)) {
gkyl_gyrokinetic_app_cout(app, stdout, "Taking time-step %ld at t = %g ...", step, t_curr);
struct gkyl_update_status status = gkyl_gyrokinetic_update(app, dt);
gkyl_gyrokinetic_app_cout(app, stdout, " dt = %g\n", status.dt_actual);
if (!status.success) {
gkyl_gyrokinetic_app_cout(app, stdout, "** Update method failed! Aborting simulation ....\n");
break;
}
t_curr += status.dt_actual;
dt = status.dt_suggested;
calc_integrated_diagnostics(&trig_calc_intdiag, app, t_curr, false, t_curr > t_end, status.dt_actual);
write_data(&trig_write_conf, &trig_write_phase, app, t_curr, false, t_curr > t_end);
if (dt_init < 0.0) {
dt_init = status.dt_actual;
}
else if (status.dt_actual < dt_failure_tol * dt_init) {
num_failures += 1;
gkyl_gyrokinetic_app_cout(app, stdout, "WARNING: Time-step dt = %g", status.dt_actual);
gkyl_gyrokinetic_app_cout(app, stdout, " is below %g*dt_init ...", dt_failure_tol);
gkyl_gyrokinetic_app_cout(app, stdout, " num_failures = %d\n", num_failures);
if (num_failures >= num_failures_max) {
gkyl_gyrokinetic_app_cout(app, stdout, "ERROR: Time-step was below %g*dt_init ", dt_failure_tol);
gkyl_gyrokinetic_app_cout(app, stdout, "%d consecutive times. Aborting simulation ....\n", num_failures_max);
calc_integrated_diagnostics(&trig_calc_intdiag, app, t_curr, false, true, status.dt_actual);
write_data(&trig_write_conf, &trig_write_phase, app, t_curr, false, true);
break;
}
}
else {
num_failures = 0;
}
step += 1;
}
gkyl_gyrokinetic_app_stat_write(app);
// Fetch simulation statistics.
struct gkyl_gyrokinetic_stat stat = gkyl_gyrokinetic_app_stat(app);
gkyl_gyrokinetic_app_cout(app, stdout, "\n");
gkyl_gyrokinetic_app_cout(app, stdout, "Number of update calls %ld\n", stat.nup);
gkyl_gyrokinetic_app_cout(app, stdout, "Number of forward-Euler calls %ld\n", stat.nfeuler);
gkyl_gyrokinetic_app_cout(app, stdout, "Number of RK stage-2 failures %ld\n", stat.nstage_2_fail);
if (stat.nstage_2_fail > 0) {
gkyl_gyrokinetic_app_cout(app, stdout, " Max rel dt diff for RK stage-2 failures %g\n", stat.stage_2_dt_diff[1]);
gkyl_gyrokinetic_app_cout(app, stdout, " Min rel dt diff for RK stage-2 failures %g\n", stat.stage_2_dt_diff[0]);
}
gkyl_gyrokinetic_app_cout(app, stdout, "Number of RK stage-3 failures %ld\n", stat.nstage_3_fail);
gkyl_gyrokinetic_app_cout(app, stdout, "Number of write calls %ld\n", stat.n_io);
gkyl_gyrokinetic_app_print_timings(app, stdout);
freeresources:
// Free resources after simulation completion.
gkyl_gyrokinetic_app_release(app);
gkyl_gyrokinetic_comms_release(comm);
#ifdef GKYL_HAVE_MPI
if (app_args.use_mpi)
MPI_Finalize();
#endif
return 0;
}
Unless you are running Flan on the same machine as Gkeyll, you will want to copy over a number of files to the machine Flan will be run on. These files include (X indicates all of the numbered files):
- d3d-167196-v9-field_X.gkyl
The plasma potential, used for calculating Electric field components
- d3d-167196-v9-elc_BiMaxwellianMoments_X.gkyl
The electron moments (density and perpendicular/parallel temperatures and flows)
- d3d-167196-v9-ion_BiMaxwellianMoments_X.gkyl
The deuterium moments (density and perpendicular/parallel temperatures and flows)
- d3d-167196-v9-bcart.gkyl
Cartesian components of magentic field (not actually used anymore in Flan)
- d3d-167196-v9-b_i.gkyl
Covariant components of magnetic field vector
- d3d-167196-v9-bmag.gkyl
Magnetic field magnitude
- d3d-167196-v9-jacobgeo.gkyl
Jacobian for coordinate transformation between computational field-aligned coordinated are Cartesian coordinates
- d3d-167196-v9-gij.gkyl
Metric coefficients
- d3d-167196-v9-dzdx.gkyl
Dual-basis vectors
- d3d-167196-v9.c
Not needed, but good practice to include input file with things for reference
Note you do NOT need the distribution function files (d3d-167196-v9-elc_X.gkyl) files. Nothing bad will happen if you copy those over, but they can take up an enormous amount of space.
Store these files all in the same directory, for this guide we will call this directory /home/zamp/gkyldir/d3d-167196-v9 just for the sake of calling it something.
Simulating Tungsten Transport with Flan
Now that we have run Gkeyll and copied over the required files, we are ready to run Flan. If you have not already, see Flan installation instructions before continuing.
First, make sure you have the (flan) conda environment active. This is installed automatically as part of the installation process, and you will know it is active if you see (flan) at the start of your terminal. If you don’t, activate it with conda activate flan. You must have the :literal:`(flan)` environment active to run Flan because it contains paths to some python scripts that serve as the interface between Flan and the Gkeyll postprocessor postgkyl.
It is useful to create a directory to contain all your Flan cases, it can be anywhere (don’t put it in the repository directory, bad practice). For this section we will assume it is called /home/zamp/flandir. Within flandir, you must use the following script to setup a simulation directory named diiid-farsol-w-v1 (the Flan repository is assumed to be located at /home/zamp/github/flan):
(flan) $ cd /home/zamp/flandir
(flan) $ /home/zamp/github/flan/scripts/flan_setup.sh diiid-farsol-w-v1
This will create a directory called diiid-farsol-w-v1 and place the needed files in it. The input file is diiid-farsol-w-v1.cpp. If you open this up, you will notice a function mapc2p, a function called create_inputs and main. At this point we will describe the high-level overview of how you, the user, interact with and run Flan.
The input file, diiid-farsol-w-v1.cpp, is actually a C++ source file. When you run make in the run directory, the Makefile that is there links compiles your input file and links it to the Flan shared library located, in this example, at /home/zamp/github/flan/lib/libflan.so. It then creates an executable appropriately named diiid-farsol-w-v1 in your directory. You then run this executable according to the rules of the machine you are on (e.g., straight from the terminal or with a slurm bash script).
If you look at the bottom of the input file, you will notice:
int main()
{
// Run Flan
Inputs inpts {create_inputs()};
flan(inpts);
}
This creates an Inputs object out of your input options and then passes it into the top-level run command for Flan. The executable you create will start here, just like any normal C++ program. In a sense, the input file is part of Flan since it all gets compiled together. The reason for this, over a normal text-based input file, is not immediately apparent from this example. The one-liner is that it enables extreme flexiblity with regards to coupling to Gkeyll from a developer perspective, a code which itself is under active development and continues to undergo major upgrades/modifications. This type of input file allows Flan to remain nimble in that regard.
This is where all the input options are entered. You will notice some input options are provided to get you started. It is good to be familiar with the Gkeyll simulation you are running flan with so that you can tell it where the impurities start. A possible bare-minimum set of input options could look like this:
inpts["case_name"] = "test";
inpts["imp_num"] = 10000;
// Tell Flan where to find the Gkeyll files and how many to use. This is one
// that used the simple helical approximation, so the coordinates are already
// Cartesian (this means nothing needs to be done with mapc2p).
inpts["gkyl_dir"] = "/home/zamp/gkyldir/d3d-167196-v6-gpu";
inpts["gkyl_casename"] = "d3d-167196-v6-gpu";
inpts["gkyl_frame_start"] = 600;
inpts["gkyl_frame_end"] = 699;
inpts["gkyl_elec_name"] = "elc";
inpts["gkyl_ion_name"] = "ion";
// Setup simulation to follow W ions
inpts["imp_num"] = 10000
inpts["imp_mass_amu"] = 183.84;
// I know from this Gkeyll simulation that this corresponds to the "left"
// or inner edge of the simulation, so start W ions there and we will
// watch them transport from there.
inpts["imp_xmin"] = 2.315;
inpts["imp_xmax"] = 2.315;
// Randomly start the ions anywhere in the y direction
inpts["imp_ystart_opt"] = "range";
// Need to know where to find the ADAS files and what year to use
inpts["openadas_root"] = "/home/zamp/flandir/openadas";
inpts["openadas_year"] = 50;
You can leave the rest of the input file alone for now. To run the simulation, you must compile it first to create an executable, then run the executable. This is easily done (don’t forget to make sure the flan conda environment is active):
(flan) $ make
(flan) $ ./test
Once finished, we can plot the data using the provided flan_plots python plotting library. Note: You must have the flan conda environment active
(flan) $ ipython
In [1]: import flan_plots
In [2]: fp = flan_plots.FlanPlots("test.nc")
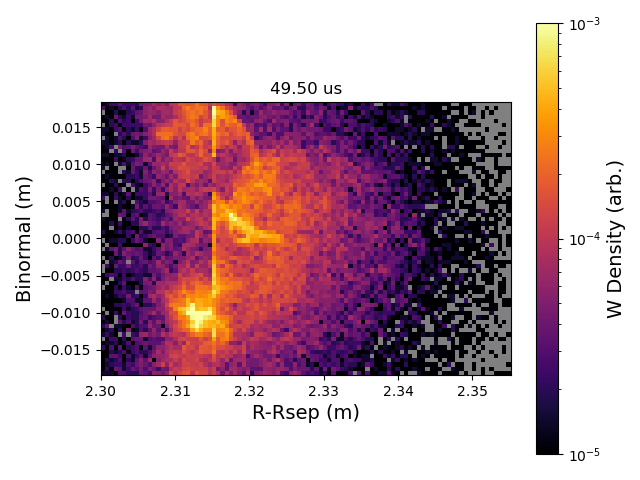
In [3]: fp.plot_frames_xy("imp_density", 0, 99, 0.0, animate_cbar=True, vmin=1e-6, vmax=1e-3, norm_type="log", xlabel="R-Rsep (m)", ylabel="Binormal (m)", cbar_label="W Density (arb.)")
This creates an animated plot, a screenshot of which is shown below.
Data can be extracted for more detailed analysis. This will be covered in future flan_plots documentation (one day).